The first objective of the study is based on experimental characterization of the studied compound. The synthetization process of C11H8O4 (I) involved the O-acetylation of 6-hydroxycoumarin with acetic anhydride, utilizing diethyl ether as a solvent and pyridine as a base. The obtained structure was characterized by both spectroscopic analyses such as ESI-MS, FT-IR, 1H and 13C NMR analysis and by single-crystal X-ray diffraction studies. In the latter case, we employed direct methods to solve the structure of (I) and subsequently refined to a final R value of 0.054 for 1896 independent reflections. In the structure, C—H•••O hydrogen bonds connect the molecules into R22 (8) dimers, which are linked together by C—H•••O interactions, forming layers parallel to the bc crystallographic plane. Similarly, the crystal structure is sustained by π–π interactions between neighboring rings, with inter-centroid distances lower than 3.8 Å. The second objective of the study is to use theoretical calculation methods to analyze the effect of solvent polarity on the energy gap of the boundary molecular orbitals and the overall chemical reactivity of coumarin-6-yl acetate in order to provide a better understanding of stability and reactivity. A series of density functional theory computations were achieved with B3LYP/6-311++G(d,p) basis set in both gas and solvent phases. In addition to the dipole moment, the natural bond orbital charge distribution was estimated in toluene, tetrahydrofuran (THF) and benzene solvents. The calculations were conducted utilizing the Gaussian 09 software, and the outcomes exhibited that the solvents have an influence on the optimized parameters. Furthermore, dual and local reactivity indices as Fukui functions from the natural bond orbital (NBO) charges were estimated in order to have a better comprehension of the electrophilic and nucleophilic regions, as well as the chemical activity of (I). The obtained dipole moment in the gas phase is 6.03 Debye and those in the presence of the solvents are 7.89, 6.87, 7.51 and 6.83 Debye for water, toluene, THF and benzene, respectively. Additionally, the global chemical reactivity parameters exhibit variation contingent on the molecular compound and polarity of the solvents, making this an important consideration in the selection of appropriate solvents for a given chemical reaction. The studied compound shows higher stability in the benzene solvent evidenced by an EHOMO-ELUMO energy gap of 9.48 eV, while its low stability is observed in the gas phase with an EHOMO-ELUMO energy gap of 6.64 eV.
| Published in | Science Journal of Chemistry (Volume 13, Issue 1) |
| DOI | 10.11648/j.sjc.20251301.12 |
| Page(s) | 11-32 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2025. Published by Science Publishing Group |
Coumarin Ester, Crystal Structure, Spectroscopic Analysis, Quantum Chemical Calculations, Fukui Functions
chemical formula | C11H8O4 | Theta range for data collection [°] | 7.1 - 76.2 |
Formula weight | 204.17 | Crystal size [mm3] | 0.28× 0.28 × 0.04 |
Temperature [K] | 298 | Index ranges | -4 ≤ h ≤4; -46 ≤ k ≤ 47; -8 ≤ l ≤ 8 |
Wavelength λ [Å] | 1.54184 | Reflections collected | 8017 |
Crystal system | Monoclinic | Absorption coefficient [mm-1] | 0.96 |
Space group | P21/n | Theta full [°] | 67.684 |
Unit cell dimensions | F(000) | 424 | |
a [Å] | 3.9039(2) | Refinement method | Full-matrix least squares on F2 |
b [Å] | 37.5379(12) | Data/restraints/parameters | 1712/0/ 136 |
c [Å] | 6.4621(3) | Goodness of fit | 1.03 |
α [°] | 90 | Final R indices [F2 > 2.0 σ(F2)] | R1 = 0.055, wR1= 0.164 |
β [°] | 103.726(4) | Density calculated [g.cm-3] | 1.265 |
γ [°] | 90 | Independent reflections | 1896 |
Volume [Å3] | 919.94(7) | Rint | 0.032 |
Z | 4 | R indices (all data) | 0.0574 |
Crystal description | plate | Δρmax, Δρmin (e Å−3) | 0.28, -0.17 |
crystal color | Colorless | (Δ/σ)max | < 0.001 |
Diffractometer | SuperNova, Dual, Cu at zero, AtlasS2 | Absorption correction | multi-scan; CrysAlisPro 1.171.42.79a (Rigaku Oxford Diffraction, 2022) Empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm. |
D—H…A | D—H | H…A | D…A | D—H…A |
|---|---|---|---|---|
C2—H5…O2i | 0.93 | 2.51 | 3.397 (2) | 160 |
C11—H11C…O4ii | 0.96 | 2.58 | 3.378 (2) | 141 |
Cg(I) | Cg(J) | Symmetry Cg(J) | Cg(I)…Cg(J) | CgI_Perp | CgJ_Perp | Slippage |
|---|---|---|---|---|---|---|
Cg1 | Cg3 | 1+x, y, z | 3.7192(8) | -3.4080 (6) | 3.4147 (5) | 1.474 |
Cg2 | Cg3 | -1+x, y, z | 3.7226 | 3.4061 | -3.4004 | 1.515 |
Atom | x | y | z | Ueq |
|---|---|---|---|---|
O1 | 0.5293 (3) | 0.08275 (3) | 0.03878 (15) | 0.0436 (3) |
O2 | 0.6298 (4) | 0.03526 (3) | −0.13667 (18) | 0.0603 (4) |
O3 | 0.7013 (3) | 0.79975 (19) | 0.76450 (16) | 0.0434 (3) |
O4 | 0.61611 (4) | 0.21027 (3) | 0.6250 (2) | 0.0677 (4) |
C1 | 0.6875 (4) | 0.04991 (4) | 0.0338 (2) | 0.0429 (4) |
C2 | 0.9057 (4) | 0.03680 (4) | 0.2330 (2) | 0.0446 (4) |
C3 | 0.9557 (4) | 0.05577 (4) | 0.4134 (2) | 0.0415 (4) |
C4 | 0.7911 (4) | 0.09011 (3) | 0.4138 (2) | 0.0348 (3) |
C5 | 0.5812 (4) | 0.10251 (3) | 0.2228 (2) | 0.0360 (3) |
C6 | 0.4105 (4) | 0.13520 (4) | 0.2078 (2) | 0.0420 (4) |
C7 | 0.4547 (4) | 0.15620 (4) | 0.3872 (2) | 0.0414 (4) |
C8 | 0.6686 (4) | 0.14433 (3) | 0.5779 (2) | 0.0367 (3) |
C9 | 0.8354 (4) | 0.11183 (4) | 0.5949 (2) | 0.0376 (3) |
C10 | 0.8350 (4) | 0.19810 (4) | 0.7676 (2) | 0.0424 (4) |
C11 | 0.8240 (5) | 0.21674 (5) | 0.9701 (3) | 0.0575 (5) |
Bond | X-Ray | Calc.(gas) | Bond | X-Ray | Calc.(gas) | Bond | X-Ray | Calc.(gas) |
|---|---|---|---|---|---|---|---|---|
O1—C5 | 1.375 (2) | 1.366 | C5—C4 | 1.390 (2) | 1.404 | C8—C9 | 1.375 (2) | 1.380 |
O1—C1 | 1.383 (2) | 1.391 | C4—C9 | 1.403 (2) | 1.406 | C9—C4 | 1.403 (2) | 1.406 |
O3—C10 | 1.358 (2) | 1.374 | C3—C2 | 1.340 (2) | 1.350 | O2—C1 | 1.204 (2) | 1.207 |
O4—C10 | 1.192 (2) | 1.202 | C4—C3 | 1.441 (2) | 1.440 | C10—C11 | 1.494 (2) | 1.502 |
O3—C8 | 1.407 (2) | 1.398 | C6—C7 | 1.379 (2) | 1.387 | C1—C2 | 1.451 (2) | 1.456 |
C5—C6 | 1.389 (2) | 1.394 | C7—C8 | 1.388 (2) | 1.397 |
Bond angle | X-Ray | Calc.(Gas) | Bond angle | X-Ray | Calc.(Gas) | Bond angle | X-Ray | Calc.(Gas) |
|---|---|---|---|---|---|---|---|---|
C5—O1—C1 | 121.72 (11) | 122.85 | O1—C5—C4 | 121.53 (12) | 121.37 | C2—C3—C4 | 120.19 (13) | 120.71 |
C10—O3—C8 | 117.99 (11) | 119.20 | C6—C5—C4 | 121.99 (13) | 121.10 | O2—C1—O1 | 116.16 (14) | 115.87 |
C8—C9—C4 | 119.07 (12) | 119.82 | C6—C7—C8 | 119.46 (13) | 119.75 | O2—C1—C2 | 126.91 (14) | 126.22 |
C5—C4—C9 | 118.47 (13) | 118.72 | C3—C2—C1 | 121.76 (14) | 121.76 | O1—C1—C2 | 116.93 (13) | 115.87 |
C5—C4—C3 | 117.86 (12) | 117.44 | C9—C8—C7 | 122.06 (13) | 121.12 | O4—C10—O3 | 123.35 (14) | 123.52 |
C9—C4—C3 | 123.67 (12) | 123.83 | C9—C8—O3 | 117.82 (12) | 117.67 | O4—C10—C11 | 125.86 (15) | 125.72 |
C7—C6—C5 | 118.95 (13) | 119.49 | C7—C8—O3 | 119.99 (13) | 121.09 | O3—C10—C11 | 110.79 (13) | 110.76 |
O1—C5—C6 | 116.48 (12) | 117.53 |
Dihedral angles | X-Ray | Calc.(Gas) | Dihedral angles | X-Ray (I) | Calc.(Gas) |
|---|---|---|---|---|---|
C8—C9—C4—C5 | 0.5 (2) | 0.17 | C6—C7—C8—C9 | −1.0 (2) | -0.23 |
C8—C9—C4—C3 | 179.43 (13) | -179.89 | C6—C7—C8—O3 | −176.70 (13) | -176.10 |
C1—O1—C5—C6 | 179.51 (13) | -179.97 | C10—O3—C8—C9 | 123.21 (15) | 120.51 |
C1—O1—C5—C4 | −1.0 (2) | -0.090 | C10—O3—C8—C7 | −60.88 (19) | -63.48 |
C7—C6—C5—O1 | −179.67 (13) | 179.82 | C1—C2—C3—C4 | 0.1 (2) | -0.08 |
C7—C6—C5—C4 | 0.9 (2) | -0.06 | C5—C4—C3—C2 | 0.1 (2) | 0.05 |
C9—C4—C5—O1 | 179.35 (12) | 179.98 | C9—C4—C3—C2 | −178.86 (14) | -179.89 |
C3—C4—C5—O1 | 0.3 (2) | 0.04 | C5—O1—C1—O2 | −178.57 (14) | -179.99 |
C9—C4—C5—C6 | −1.2 (2) | -0.15 | C5—O1—C1—C2 | 1.2 (2) | 0.05 |
C3—C4—C5—C6 | 179.74 (13) | 179.91 | C3—C2—C1—O2 | 178.99 (17) | -179.91 |
C5—C6—C7—C8 | 0.2 (2) | 0.25 | C3—C2—C1—O1 | −0.8 (2) | 0.04 |
C4—C9—C8—C7 | 0.6 (2) | 0.02 | C8—O3—C10—O4 | −4.4 (2) | -0.41 |
C4—C9—C8—O3 | 176.43 (12) | 176.03 | C8—O3—C10—C11 | 175.44 (13) | 179.69 |
Gas | Water | THF | Benzene | Toluene | |
|---|---|---|---|---|---|
ELUMO (eV) | -1.48 | -1.51 | 0.74 | 0.75 | -1.51 |
EHOMO (eV) | -8.12 | -8.24 | -8.72 | -8.73 | -8.20 |
I (eV) | 8.12 | 8.24 | 8.72 | 8.73 | 8.20 |
A (eV) | 1.48 | 1.51 | -0.74 | -0.75 | 1.51 |
𝜒 (eV) | 4.80 | 4.88 | 3.99 | 3.99 | 4.86 |
(eV) | -4.80 | -4.88 | -3.99 | -3.99 | -4.86 |
𝜂 (eV) | 3.32 | 3.37 | 4.73 | 4.74 | 3.35 |
(eV-1) | 0.151 | 0.149 | 0.106 | 0.105 | 0.149 |
ω (eV) | 3.470 | 3.531 | 1.683 | 1.679 | 3.523 |
ΔEg (eV) | 6.64 | 6.73 | 9.46 | 9.48 | 6.69 |
Atoms | (I) in gas phase | (I) in water | (I) in tetrahydrofuran (THF) | ||||||
|---|---|---|---|---|---|---|---|---|---|
q(N) | q(N-1) | q(N+1) | q(N) | q(N-1) | q(N+1) | q(N) | q(N-1) | q(N+1) | |
O1 | -0.53336 | -0.46999 | -0.31181 | -0.53639 | -0.20537 | -0.32012 | -0.53563 | -0.47618 | -0.31842 |
O2 | -0.56065 | -0.39709 | -0.38437 | -0.63017 | -0.13050 | -0.41971 | -0.61698 | -0.45751 | -0.41318 |
O3 | -0.58162 | -0.55533 | -0.29229 | -0.57929 | -0.27335 | -0.29221 | -0.58658 | -0.57051 | -0.29603 |
O4 | -0.57973 | -0.55064 | -0.29714 | -0.62658 | -0.30323 | -0.31862 | -0.61039 | -0.59029 | -0.31042 |
C1 | 0.77962 | 0.74965 | 0.34921 | 0.79262 | 0.34976 | 0.32145 | 0.79033 | 0.76789 | 0.32745 |
C2 | -0.30309 | -0.18428 | -0.34109 | -0.31474 | 0.04652 | -0.31964 | -0.31245 | -0.16755 | -0.32442 |
C3 | -0.11439 | -0.12996 | -0.25830 | -0.09152 | -0.09878 | -0.29738 | -0.09644 | -0.10713 | -0.29016 |
C4 | -0.14627 | -0.04456 | -0.06660 | -0.14306 | 0.08937 | -0.05036 | -0.14343 | -0.02250 | -0.05277 |
C5 | 0.35491 | 0.47528 | 0.12460 | 0.35158 | 0.36138 | 0.10857 | 0.35223 | 0.48672 | 0.11184 |
C6 | -0.22437 | -0.21621 | -0.13673 | -0.22777 | -0.12955 | -0.12445 | -0.22677 | -0.22181 | -0.12674 |
C7 | -0.19454 | -0.12251 | -0.26101 | -0.19893 | 0.00942 | -0.24761 | -0.19814 | -0.10453 | -0.25041 |
C8 | 0.28574 | 0.43492 | 0.16657 | 0.28051 | 0.30544 | 0.16885 | 0.28301 | 0.41843 | 0.16947 |
C9 | -0.19124 | -0.21069 | -0.21340 | -0.18776 | -0.14883 | -0.21011 | -0.18824 | -0.20544 | -0.21095 |
C10 | 0.83414 | 0.83262 | 0.41574 | 0.85488 | 0.43029 | 0.42756 | 0.85026 | 0.85070 | 0.42499 |
C11 | -0.68336 | -0.68734 | -0.33992 | -0.68700 | -0.34308 | -0.34353 | -0.68512 | -0.68437 | -0.34249 |
Atoms | (I) in benzene | (I) in toluene | ||||
|---|---|---|---|---|---|---|
q(N) | q(N-1) | q(N+1) | q(N) | q(N-1) | q(N+1) | |
O1 | -0.53436 | -0.19076 | -0.31548 | -0.60952 | -0.19175 | -0.58218 |
O2 | -0.59137 | -0.10870 | -0.40012 | -0.69016 | -0.09898 | -0.58218 |
O3 | -0.58792 | -0.27062 | -0.29643 | -0.65029 | -0.27263 | -0.58575 |
O4 | -0.59214 | -0.28372 | -0.30178 | -0.69248 | -0.29096 | -0.61261 |
C1 | 0.78555 | 0.33982 | 0.33826 | 0.92877 | 0.33413 | 0.74432 |
C2 | -0.30818 | 0.02414 | -0.33297 | -0.34388 | 0.03413 | -0.45571 |
C3 | -0.10521 | -0.10344 | -0.27590 | -0.03853 | -0.10174 | -0.28178 |
C4 | -0.14464 | 0.07342 | -0.05837 | -0.17075 | 0.07665 | -0.12881 |
C5 | 0.35367 | 0.35840 | 0.11716 | 0.40352 | 0.35440 | 0.31055 |
C6 | -0.22525 | -0.11956 | -0.13078 | -0.22442 | -0.12234 | -0.25998 |
C7 | -0.19600 | -0.00791 | -0.25553 | -0.16227 | -0.00285 | -0.32170 |
C8 | 0.28538 | 0.32426 | 0.16892 | 0.30069 | 0.31554 | 0.28041 |
C9 | -0.18950 | 0.32426 | -0.21160 | -0.15429 | -0.15038 | -0.27529 |
C10 | 0.84209 | 0.42322 | 0.42038 | 0.97617 | 0.42385 | 0.83905 |
C11 | -0.68346 | -0.34198 | -0.34129 | -0.61559 | -0.34330 | -0.68410 |
Atoms | Local descriptors | Dual descriptors | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
|
|
|
| |
O1 | -0.06337 | 0.22155 | -0.00957 | 0.03345 | -0.21039 | 0.73555 | -0.21989 | 0.76878 | 0.28492 | 0.04302 | 0.98867 |
O2 | -0.16356 | 0.17628 | -0.02470 | 0.02662 | -0.54302 | 0.58525 | -0.56755 | 0.61169 | 0.33984 | 0.05132 | 1.17924 |
O3 | -0.02629 | 0.28933 | -0.00397 | 0.04369 | -0.08728 | 0.96058 | -0.09123 | 1.00398 | 0.31562 | 0.04766 | 1.09520 |
O4 | -0.02909 | 0.28259 | -0.00439 | 0.04267 | -0.09658 | 0.93820 | -0.10094 | 0.98059 | 0.31168 | 0.04706 | 1.08153 |
C1 | 0.02997 | -0.43041 | 0.00453 | -0.06499 | 0.09950 | -1.42896 | 0.10400 | -1.49352 | -0.46038 | -0.06952 | -1.59752 |
C2 | -0.11881 | -0.038 | -0.01794 | -0.00574 | -0.39445 | -0.12616 | -0.41227 | -0.13186 | 0.08081 | 0.01220 | 0.28041 |
C3 | 0.01557 | -0.14391 | 0.00235 | -0.02173 | 0.05169 | -0.47778 | 0.05403 | -0.49937 | -0.15948 | -0.02408 | -0.55340 |
C4 | -0.10171 | 0.07967 | -0.01536 | 0.01203 | -0.33768 | 0.26450 | -0.35293 | 0.27645 | 0.18138 | 0.02739 | 0.62939 |
C5 | -0.12037 | -0.23031 | -0.01818 | -0.03478 | -0.39963 | -0.76463 | -0.41768 | -0.79918 | -0.10994 | -0.01660 | -0.38149 |
C6 | -0.00816 | 0.08764 | -0.00123 | 0.01323 | -0.02709 | 0.29096 | -0.02832 | 0.30411 | 0.0958 | 0.01447 | 0.33243 |
C7 | -0.07203 | -0.06647 | -0.01088 | -0.01004 | -0.23914 | -0.22068 | -0.24994 | -0.23065 | 0.00556 | 0.00084 | 0.01929 |
C8 | -0.14918 | -0.11917 | -0.02253 | -0.01799 | -0.49528 | -0.39564 | -0.51765 | -0.41352 | 0.03001 | 0.00453 | 0.10413 |
C9 | 0.01945 | -0.02216 | 0.00294 | -0.00335 | 0.06457 | -0.07357 | 0.06749 | -0.07690 | -0.04161 | -0.00628 | -0.14439 |
C10 | 0.00152 | -0.4184 | 0.00023 | -0.06318 | 0.00505 | -1.38909 | 0.00527 | -1.45185 | -0.41992 | -0.06341 | -1.45712 |
C11 | 0.00398 | 0.34344 | 0.00060 | 0.05186 | 0.01321 | 1.14022 | 0.01382 | 1.19172 | 0.33946 | 0.05126 | 1.17790 |
Atoms | Local descriptors | Dual descriptors | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
|
|
|
| |
O1 | -0.33102 | 0.21627 | -0.04932 | 0.03222 | -1.11554 | 0.72883 | -1.16883 | 0.76365 | 0.54729 | 0.08155 | 1.93248 |
O2 | -0.49967 | 0.21046 | -0.07445 | 0.03136 | -1.68389 | 0.70925 | -1.76433 | 0.74313 | 0.71013 | 0.10581 | 2.50747 |
O3 | -0.30594 | 0.28708 | -0.04559 | 0.04277 | -1.03102 | 0.96746 | -1.08027 | 1.01368 | 0.59302 | 0.08836 | 2.09395 |
O4 | -0.32335 | 0.30796 | -0.04818 | 0.04589 | -1.08969 | 1.03783 | -1.14175 | 1.08741 | 0.63131 | 0.09407 | 2.22916 |
C1 | 0.44286 | -0.47117 | 0.06599 | -0.07020 | 1.49244 | -1.58784 | 1.56374 | -1.66370 | -0.91403 | -0.13619 | -3.22744 |
C2 | -0.36126 | -0.0049 | -0.05383 | -0.00073 | -1.21745 | -0.01651 | -1.27561 | -0.01730 | 0.35636 | 0.05310 | 1.25831 |
C3 | 0.00726 | -0.20586 | 0.00108 | -0.03067 | 0.02447 | -0.69375 | 0.02564 | -0.72689 | -0.21312 | -0.03175 | -0.75253 |
C4 | -0.23243 | 0.0927 | -0.03463 | 0.01381 | -0.78329 | 0.31240 | -0.82071 | 0.32732 | 0.32513 | 0.04844 | 1.14803 |
C5 | -0.0098 | -0.24301 | -0.00146 | -0.03621 | -0.03303 | -0.81894 | -0.03460 | -0.85807 | -0.23321 | -0.03475 | -0.82346 |
C6 | -0.09822 | 0.10332 | -0.01463 | 0.01539 | -0.33100 | 0.34819 | -0.34681 | 0.36482 | 0.20154 | 0.03003 | 0.71164 |
C7 | -0.20835 | -0.04868 | -0.03104 | -0.00725 | -0.70214 | -0.16405 | -0.73568 | -0.17189 | 0.15967 | 0.02379 | 0.56379 |
C8 | -0.02493 | -0.11166 | -0.00371 | -0.01664 | -0.08401 | -0.37629 | -0.08803 | -0.39427 | -0.08673 | -0.01292 | -0.30624 |
C9 | -0.03893 | -0.02235 | -0.00580 | -0.00333 | -0.13119 | -0.07532 | -0.13746 | -0.07892 | 0.01658 | 0.00247 | 0.05854 |
C10 | 0.42459 | -0.42732 | 0.06326 | -0.06367 | 1.43087 | -1.44007 | 1.49923 | -1.50887 | -0.85191 | -0.12693 | -3.00809 |
C11 | -0.34392 | 0.34347 | -0.05124 | 0.05118 | -1.15901 | 1.15749 | -1.21438 | 1.21279 | 0.68739 | 0.10242 | 2.42717 |
Atoms | Local descriptors | Dual descriptors | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
|
|
|
| |
O1 | -0.41777 | 0.02734 | -0.06225 | 0.00407 | -1.39953 | 0.09159 | -1.47180 | 0.09632 | 0.44511 | 0.06632 | 1.56812 |
O2 | -0.59118 | 0.10798 | -0.08809 | 0.01609 | -1.98045 | 0.36173 | -2.08273 | 0.38041 | 0.69916 | 0.10417 | 2.46314 |
O3 | -0.37766 | 0.06454 | -0.05627 | 0.00962 | -1.26516 | 0.21621 | -1.33050 | 0.22737 | 0.4422 | 0.06589 | 1.55787 |
O4 | -0.40152 | 0.07987 | -0.05983 | 0.01190 | -1.34509 | 0.26756 | -1.41455 | 0.28138 | 0.48139 | 0.07173 | 1.69594 |
C1 | 0.59464 | -0.18445 | 0.08860 | -0.02748 | 1.99204 | -0.61791 | 2.09492 | -0.64982 | -0.77909 | -0.11608 | -2.74473 |
C2 | -0.37801 | -0.11183 | -0.05632 | -0.01666 | -1.26633 | -0.37463 | -1.33173 | -0.39398 | 0.26618 | 0.03966 | 0.93775 |
C3 | 0.06321 | -0.24325 | 0.00942 | -0.03624 | 0.21175 | -0.81489 | 0.22269 | -0.85697 | -0.30646 | -0.04566 | -1.07966 |
C4 | -0.2474 | 0.04194 | -0.03686 | 0.00625 | -0.82879 | 0.14050 | -0.87159 | 0.14775 | 0.28934 | 0.04311 | 1.01934 |
C5 | 0.04912 | -0.09297 | 0.00732 | -0.01385 | 0.16455 | -0.31145 | 0.17305 | -0.32753 | -0.14209 | -0.02117 | -0.50058 |
C6 | -0.10208 | -0.03556 | -0.01521 | -0.00530 | -0.34197 | -0.11913 | -0.35963 | -0.12528 | 0.06652 | 0.00991 | 0.23435 |
C7 | -0.15942 | -0.15943 | -0.02375 | -0.02376 | -0.53406 | -0.53409 | -0.56164 | -0.56167 | -1E-05 | 0.00000 | -0.00004 |
C8 | -0.01485 | -0.02028 | -0.00221 | -0.00302 | -0.04975 | -0.06794 | -0.05232 | -0.07145 | -0.00543 | -0.00081 | -0.01913 |
C9 | -0.00391 | -0.121 | -0.00058 | -0.01803 | -0.01310 | -0.40535 | -0.01377 | -0.42628 | -0.11709 | -0.01745 | -0.41251 |
C10 | 0.55232 | -0.13712 | 0.08230 | -0.02043 | 1.85027 | -0.45935 | 1.94582 | -0.48307 | -0.68944 | -0.10273 | -2.42890 |
C11 | -0.27229 | -0.06851 | -0.04057 | -0.01021 | -0.91217 | -0.22951 | -0.95928 | -0.24136 | 0.20378 | 0.03036 | 0.71792 |
T(K) | S | Cp | H | G |
|---|---|---|---|---|
25 | 55.757 | 11.433 | 103.617 | 102.223 |
50 | 65.087 | 15.67 | 103.959 | 100.705 |
75 | 72.048 | 18.801 | 104.391 | 98.987 |
100 | 77.864 | 21.798 | 104.898 | 97.112 |
125 | 83.064 | 24.956 | 105.482 | 95.099 |
150 | 87.907 | 28.308 | 106.148 | 92.962 |
175 | 92.533 | 31.822 | 106.899 | 90.706 |
200 | 97.018 | 35.452 | 107.74 | 88.336 |
225 | 101.407 | 39.149 | 108..672 | 85.856 |
250 | 105..724 | 42.868 | 109.697 | 83.266 |
275 | 109.984 | 46.563 | 110.815 | 80.57 |
298.15 | 113.882 | 49.932 | 111.932 | 77.768 |
300 | 114.192 | 50.198 | 112.025 | 77.768 |
325 | 118.35 | 53.741 | 113.324 | 74.861 |
350 | 122.459 | 57.169 | 114.711 | 71.85 |
375 | 126.516 | 60.463 | 116.182 | 68.738 |
400 | 130.52 | 63.613 | 117.733 | 65.525 |
425 | 134.467 | 66.614 | 119.361 | 62.213 |
450 | 138.356 | 69.464 | 121.063 | 58.802 |
475 | 142.185 | 72.165 | 122.833 | 55.295 |
500 | 145.952 | 74.721 | 124.67 | 51.693 |
525 | 149.657 | 77.138 | 126.568 | 47.998 |
550 | 153.299 | 79.422 | 128.525 | 44.211 |
575 | 156.877 | 81.582 | 130.538 | 40.334 |
600 | 160.393 | 83.624 | 132.603 | 36.368 |
625 | 163.846 | 85.556 | 134.718 | 32.315 |
650 | 167.238 | 87.385 | 136.88 | 28.176 |
675 | 170.568 | 89.119 | 139.087 | 23.953 |
700 | 173.839 | 90.763 | 141.336 | 19.648 |
725 | 177.052 | 92.323 | 143.624 | 15.262 |
750 | 180.207 | 93.806 | 145.951 | 10.796 |
775 | 183.306 | 95.216 | 148.314 | 6.252 |
800 | 186.35 | 96.557 | 150.711 | 1.631 |
825 | 189.341 | 97.835 | 153.141 | -3.065 |
850 | 192.28 | 99.053 | 155.603 | -7.836 |
875 | 195.168 | 100.215 | 158.094 | -12.679 |
900 | 198.007 | 101.324 | 160.613 | -17.594 |
925 | 200.798 | 102.383 | 163.159 | -22.579 |
950 | 203.542 | 103.395 | 165.732 | -27.633 |
975 | 206.24 | 104.362 | 168.329 | -32.755 |
1000 | 208.894 | 105.288 | 170.949 | -37.945 |
ESI-MS | ElectroSpray Ionization Mass Spectrometry |
FT-IR | Fourier Transform Infrared Spectroscopy |
1H NMR | Proton Nuclear Magnetic Resonance |
13C NMR | Carbon-13 (C13) Nuclear Magnetic Resonance |
B3LYP | Becke, 3-parameter, Lee–Yang–Parr |
THF | TetraHydroFuran |
NBO | Natural Bond Orbital |
HOMO | Highest Occupied Molecular Orbital |
LUMO | Lowest unoccupied molecular orbital |
CFIA | Canadian Food Inspection Agency |
ADI | Acceptable Daily Intake |
EFSA | European Food Safety Authority |
NLO | Non-Linear Optical |
XRD | X-ray Diffraction |
3D | Three-Dimensional |
FMO | Frontier Molecular Orbitals |
FF | Fukui Function |
PWC | Perdew-Wang |
DND | Double-numerical + d |
TMS | Tetramethylsilane |
DFT | Density Functional Theory |
HSQC | Heteronuclear Single-Quantum Correlation |
ATR | Attenuated Total Reflectance |
APT | Attached Proton Test |
CDCl3 | Deuterated Chloroform |
RMSD | Root Mean Square Deviations |
MEP | Molecular Electrostatic Potential |
| [1] | Matos, M. J., Santana, L., Uriarte, E., Abreu, O. A., Molina, E., & Yordi, E. G. (2015). Coumarins — An Important Class of Phytochemicals. Phytochemicals - Isolation, Characterisation and Role in Human Health. (pp. 113-140). Royaune-Uni: IntechOpen. |
| [2] | Zeng, L., Zhang, R.-Y., Meng, T., Lou, Z.-C. (1990). Determination of nine flavonoids and coumarins in licorice root by high-performance liquid chromatography. J. Chromatogr. A, 513, 247-254. |
| [3] | Shojaii, A., Fard, M. H., 2012. Review of pharmacological properties and chemical constituents of Pimpella anisum. ISRN Pharmaceutics, 2012, 510795. |
| [4] | Abraham, K., Wöhrlin, F., Lindtner, O., Heinemeyer, G., Lampen, A., 2010. Toxicology and risk assessment of coumarin: Focus on human data. Mol. Nutr. Food Res, 54(2), 228-239. |
| [5] | Lake, B. G. (1999). Coumarin metabolism, toxicity and carcinogenicity: Relevance for human risk assessment. Food and Chemical Toxicology, 37(4), 423-453. |
| [6] | Coumarin in flavourings and other food ingredients with flavouring properties. Scientific opinion of the panel on food additives, flavourings, processing aids and materials in contact with food (AFC). (2008). EFSA Journal, 793, 1-15. |
| [7] | Basanagouda M, Kulkarni M V, Sharma D, Gupta V K, Pranesha P, Sandhyarani P and Rasal V P., (2009). J. Chem. Sci. 121, 485–495. |
| [8] | Vuković N, Sukdolak S, Solujić S and Niciforović N, (2010). An efficient synthesis and antioxidant properties of novel imino and amino derivatives of 4-hydroxy coumarins. Arch. Pharm. Res. 33; 5–15. |
| [9] | Emmanuel-Giota A A, Fylaktakidou K C, Litinas K E, Nicolaides D N and Hadjipavlou-Litina D J,. (2001). Synthesis and biological evaluation of several 3-(coumarin-4-yl) tetrahydroisoxazole and 3-(coumarin-4-yl) dihydropyrazole derivatives. Heterocycl. Chem.; 38: 717–722. |
| [10] | D. R. Kanis, M. A. Ratner, T. J. Marks, (1994). Design and construction of molecular assemblies with large second-order optical nonlinearities. Quantum chemical aspects. Chem. Rev. 94, 195–242. |
| [11] | D. J. Williams, (1992). Non-linear optical properties of organic materials. Thin Solid Films 216, 117–122 |
| [12] | P. J. Mendes, J. P. Prates Ramalho, A. J. E. Candeias, M. P. Robalo, M. H. Garcia, (2005). Density functional theory calculations on η 5 - monocyclopentadienylnitrilecobalt complexes concerning their second-order nonlinear optical properties. J. Mol. Struct. (Theochem.) 729 109–113 |
| [13] | N. M. F. S. A. Cerqueira, A. M. F. Oliveira-Campos, P. J. Coelho, L. H. Melo de Carvalho, A. Samat, R. Guglielmetti, (2005). Synthesis of Photochromic Dyes Based on Annulated Coumarin Systems. Helv. Chim. Acta 85, 442–450. ttps://doi.org/10.1002/1522-2675(200202)85:2<442::AID-HLCA442>3.0.CO;2-9 |
| [14] | S. P. G. Costa, J. Griffiths, G. Kirsch, A. M. F. Oliveira-Campos. Synthesis of thieno[2,3-d]thiazole derived dyes with potential application in nonlinear optics. Ann. Quım. Int. Ed. (1998), 94, 186–188. |
| [15] | M. M. M. Raposo, A. M. R. C. Sousa, A. M. C. Fonseca, G. Kirsch, (2005). Thienylpyrrole Azo Dyes: Synthesis, Solvatochromic and Electrochemical Properties. Tetrahedron 61, 8249–8256. |
| [16] | N. Arumugam, A. I. Almansour, R. S. Kumar, V. S. Krishna, D. Sriram, N. Dege. (2021). Stereoselective synthesis and discovery of novel spirooxindolopyrrolidine engrafted indandione heterocyclic hybrids as antimycobacterial agents. Bioorg. Chem., 110, 104798. |
| [17] | Spek, A. L., 2009. Structure validation in chemical crystallography. Acta Cryst., D65, 148–155. |
| [18] | Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., et al.; (2013) GAUSSIAN09. Gaussian, Inc., Wallingford, CT, USA. |
| [19] | B. Delley, (1990). An All-Electron Numerical Method for Solving the Local Density Functional for Polyatomic Molecules. J. Chem. Phys. 92, 508-517. |
| [20] | B. Delley, (2000). From Molecules to Solids with DMol3. J. Chem. Phys. 113, 7756-7764. |
| [21] | Tse-Lok, H., (1975). Hard soft acids bases (HSAB) principle and organic chemistry. Chem. Rev. 75(1), 1-20. |
| [22] | R. G. Pearson, (1963). Hard and Soft Acids and Bases. J. Amer. Chem. Soc, 85(22), 3533-3539. |
| [23] | Abou A, Sosso S., Kouassi A. F., Zoueu T. J., Djandé A., Ouari O. (2021) Synthesis, Characterization, Crystal Structure and Quantum Chemical Calculations of 2-oxo-2H-chromen-3-yl Acetate. Sci. J. Chem., 9(2), 29-44 |
| [24] |
Rigaku OD (2015). CrysAlis PRO. Rigaku Oxford Diffraction, Yarnton, England.
https://rigaku.com/products/crystallography/x-ray-diffraction/crysalispro |
| [25] | Burla M C, Caliandro R, Carrozzini B, Cascarano G L, Cuocci C, Giacovazzo C, Mallamo M, Mazzone A and Polidori G, (2015). Crystal structure determination and refinement via SIR2014. J. Appl. Cryst. 48, 306–309. |
| [26] | Farrugia L J., (2012). WinGX and ORTEP for Windows: An Update. J. Appl. Cryst. 45, 849–854. |
| [27] |
Sheldrick G M., (2015). Crystal Structure Refinement with SHELXL. Acta Cryst. C71, 3–8.
https://journals.iucr.org/c/issues/2015/01/00/fa3356/fa3356.pdf |
| [28] | Martinez-Araya J. I., (2014). Why is the dual descriptor a more accurate local reactivity descriptor than Fukui functions? J. Math. Chem. 53(2), 451 - 465. |
| [29] | Canevet, D., Salle, M., Zhang, G., Zhang, D., and Zhu, D., (2009). Tetrathiafulvalene (TTF) derivatives: key building-blocks for switchable processes. Chem. Comm. 17, 2245-2269. |
| [30] | Vektariene, A., Vektaris, G., and Svoboda, J., (2009). A theoretical approach to the nucleophilic behavior of benzofused thieno [3,2-b] furans using DFT and HF based reactivity descriptors, Arkivoc: Online Journal of Organic Chemistry. ARKIVOC. vii, 311-329. |
| [31] |
Islam, M. J., Kumer, A., Sarker, N., Paul, S., and Zannat, A., (2019). The prediction and theoretical study for chemical reactivity, thermophysical and biological activity of morpholinium nitrate and nitrite ionic liquid crystals: A DFT study. Adv. J. Chem. A 2(4), 316-326.
https://www.researchgate.net/publication/333081305_The_prediction_and_theoretical_study_for_chemical_reactivity_thermophysical_and_biological_activity_of_morpholinium_nitrate_and_nitrite_ionic_liquid_crystals_A_DFT_study https://doi.org/10.33945/SAMI/AJCA.2019.4.5 |
| [32] | E. AlShamaileh, (2014). DFT Study of Monochlorinated Pyrene Compounds. Comput. Chem. 2, 43-49. |
| [33] | Flores-Holguín, N., Frau, J., and Glossman-Mitnik, D., (2019). Chemical reactivity and bioactivity properties of the Phallotoxin family of fungal peptides based on Conceptual Peptidology and DFT study. Heliyon, 5(8), e02335. |
| [34] | Abbaz, T., Bendjeddou, A., and Villemin, D., (2018). Molecular structure, HOMO, LUMO, MEP, natural bond orbital analysis of benzo and anthraquinodimethane derivatives. Pharmaceutical and Biological Evaluations, 5(2), 27-39. |
| [35] | El_Kalai F., Çınar E. B., Lai C. H., Said Daoui S., Chelfi T., Allali M., Dege N., Karrouchi K., Benchat N., (2020). Synthesis, spectroscopy, crystal structure, TGA/DTA study, DFT and molecular docking investigations of (E)-4-(4-methylbenzyl) -6-styrylpyridazin-3(2H)-one, J. Mol. Struct., 129435 |
| [36] | Agrahari B., Layek S., Ganguly R, Dege N., Pathak D. D. (2019). Synthesis, characterization and single crystal X-ray studies of pincer type Ni(II)-Schiff base complexes: Application in synthesis of 2-substituted benzimidazoles. J. Organomet. Chem., 890, 13-20. |
| [37] | Abou A., Yoda J., Djandé A., Coussan S. and Zoueu T J. (2018). Crystal structure of 2-oxo-2H-chromen-7-yl 4-fluorobenzoate. Acta Cryst. E74, 761–765. |
| [38] | Abou A., Sosso S., Kouassi A F., Zoueu T J., Djandé A., Ouari O. (2021). Synthesis, Characterization, Crystal Structure and Quantum Chemical Calculations of 2-oxo-2H-chromen-3-yl Acetate. Sci. J. Chem. 9(2), 29-44 |
| [39] | Koulabiga Z., Yao K H., Abou A., Djandé A., Giorgi M., Coussan S. (2024). Synthesis, Characterization, Hirshfeld Surface Analysis and Quantum Chemical Calculations of 2-oxo-2H- Chromen-6-yl 4-Methoxybenzoate. Am. J. Org. Chem., 12(1): 1-19. |
| [40] | Janiak J, J. (2000). A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands. Chem. Soc. Dalton Trans. 3885–3896. |
| [41] | D. L. Beveridge; R. Lavery Theoretical Biochemistry and Molecular Biophysics: DNA. Proteins, Adenine Press, 1990. |
| [42] | Politzer P. and J. S. Murray J. S.; (2002). The fundamental nature and role of the electrostatic potential in atoms and molecules. Theor. Chem. Acc. 108(3), 134–142. |
| [43] | Pearson, R. G. (1986). Absolute electronegativity and hardness correlated with molecular orbital theory. Proc. Natl. Acad. Sci. U.S.A. Nov;. 83(22): 8440-8441. |
| [44] | Pearson, R. G. (1963) Hard and Soft Acids and Bases. J. Am. Chem. Soc. 85, 3533-3539. |
| [45] | Yang, W., Parr, R. G. and Pucci, R. (1984). Electron Density, Kohn-Sham Frontier Orbitals, and Fukui Functions. J. Chem. Phys. 81, 2862-2863. |
| [46] | Yang, W. and Mortier, W. (1986) The Use of Global and Local Molecular Parameters for the Analysis of the Gas-Phase Basicity of Amines. J. Am. Chem. Soc. 108, 5708-5711. |
| [47] | Yang, W. and Parr, R. G. (1985). Chemistry. Hardness, Softness, and the Fukui Function in the Electronic Theory of Metals and Catalysis. Proceedings of the National Academy of Sciences of the United States of America, 82, 6723-6726. |
| [48] | Chattaraj, P. K., Maiti, B. and Sarkar, U. (2003) Philicity: A Unified Treatment of Chemical Reactivity and Selectivity. J. Phys. Chem. A 107, 4973-4975. |
| [49] | Fuentealba, P. and Contreras, R. (2002). Fukui Function in Chemistry. In: Sen, K. D., Ed., Reviews in Modern Quantum Chemistry: A Celebration of the Contributions of Robert G Parr, World Scientific, River Edge, 1013-1052. |
| [50] | Padmanabhan, J., Parthasarathi, R., Elango, M., Subramanian, V., Krishnamoorthy, B. S., Gutierrez-Oliva, S., Toro-Labbe, A., Roy, D. R. and Chattaraj, P. K. (2007) Multiphilic Descriptor for Chemical Reactivity and Selectivity. J. Phys. Chem. 111, 9130-9138. |
| [51] | Morell, C., Grand, A. and Toro-Labbé, A. (2005). New Dual Descriptor for Chemical Reactivity. J. Phys. Chem. 109, 205-212. |
| [52] | Morell, C., Grand, A. and Toro-Labbé, A. (2006) Theoretical Support for Using the Delta f(r). Descriptor Chem. Phys. Lett. 425, 342-346. |
APA Style
Yao, H. K., Koulabiga, Z., Abou, A., Djandé, A., Coussan, S., et al. (2025). Synthesis, Experimental Characterizations and Theoretical Study of the Chemical Reactivity of Coumarin-6-yl Acetate in Gas and Solvent Phases. Science Journal of Chemistry, 13(1), 11-32. https://doi.org/10.11648/j.sjc.20251301.12
ACS Style
Yao, H. K.; Koulabiga, Z.; Abou, A.; Djandé, A.; Coussan, S., et al. Synthesis, Experimental Characterizations and Theoretical Study of the Chemical Reactivity of Coumarin-6-yl Acetate in Gas and Solvent Phases. Sci. J. Chem. 2025, 13(1), 11-32. doi: 10.11648/j.sjc.20251301.12
AMA Style
Yao HK, Koulabiga Z, Abou A, Djandé A, Coussan S, et al. Synthesis, Experimental Characterizations and Theoretical Study of the Chemical Reactivity of Coumarin-6-yl Acetate in Gas and Solvent Phases. Sci J Chem. 2025;13(1):11-32. doi: 10.11648/j.sjc.20251301.12
@article{10.11648/j.sjc.20251301.12,
author = {Honoré Kouadio Yao and Zakaria Koulabiga and Akoun Abou and Abdoulaye Djandé and Stéphane Coussan and Olivier Ouari},
title = {Synthesis, Experimental Characterizations and Theoretical Study of the Chemical Reactivity of Coumarin-6-yl Acetate in Gas and Solvent Phases},
journal = {Science Journal of Chemistry},
volume = {13},
number = {1},
pages = {11-32},
doi = {10.11648/j.sjc.20251301.12},
url = {https://doi.org/10.11648/j.sjc.20251301.12},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.sjc.20251301.12},
abstract = {The first objective of the study is based on experimental characterization of the studied compound. The synthetization process of C11H8O4 (I) involved the O-acetylation of 6-hydroxycoumarin with acetic anhydride, utilizing diethyl ether as a solvent and pyridine as a base. The obtained structure was characterized by both spectroscopic analyses such as ESI-MS, FT-IR, 1H and 13C NMR analysis and by single-crystal X-ray diffraction studies. In the latter case, we employed direct methods to solve the structure of (I) and subsequently refined to a final R value of 0.054 for 1896 independent reflections. In the structure, C—H•••O hydrogen bonds connect the molecules into R22 (8) dimers, which are linked together by C—H•••O interactions, forming layers parallel to the bc crystallographic plane. Similarly, the crystal structure is sustained by π–π interactions between neighboring rings, with inter-centroid distances lower than 3.8 Å. The second objective of the study is to use theoretical calculation methods to analyze the effect of solvent polarity on the energy gap of the boundary molecular orbitals and the overall chemical reactivity of coumarin-6-yl acetate in order to provide a better understanding of stability and reactivity. A series of density functional theory computations were achieved with B3LYP/6-311++G(d,p) basis set in both gas and solvent phases. In addition to the dipole moment, the natural bond orbital charge distribution was estimated in toluene, tetrahydrofuran (THF) and benzene solvents. The calculations were conducted utilizing the Gaussian 09 software, and the outcomes exhibited that the solvents have an influence on the optimized parameters. Furthermore, dual and local reactivity indices as Fukui functions from the natural bond orbital (NBO) charges were estimated in order to have a better comprehension of the electrophilic and nucleophilic regions, as well as the chemical activity of (I). The obtained dipole moment in the gas phase is 6.03 Debye and those in the presence of the solvents are 7.89, 6.87, 7.51 and 6.83 Debye for water, toluene, THF and benzene, respectively. Additionally, the global chemical reactivity parameters exhibit variation contingent on the molecular compound and polarity of the solvents, making this an important consideration in the selection of appropriate solvents for a given chemical reaction. The studied compound shows higher stability in the benzene solvent evidenced by an EHOMO-ELUMO energy gap of 9.48 eV, while its low stability is observed in the gas phase with an EHOMO-ELUMO energy gap of 6.64 eV.},
year = {2025}
}
TY - JOUR T1 - Synthesis, Experimental Characterizations and Theoretical Study of the Chemical Reactivity of Coumarin-6-yl Acetate in Gas and Solvent Phases AU - Honoré Kouadio Yao AU - Zakaria Koulabiga AU - Akoun Abou AU - Abdoulaye Djandé AU - Stéphane Coussan AU - Olivier Ouari Y1 - 2025/01/24 PY - 2025 N1 - https://doi.org/10.11648/j.sjc.20251301.12 DO - 10.11648/j.sjc.20251301.12 T2 - Science Journal of Chemistry JF - Science Journal of Chemistry JO - Science Journal of Chemistry SP - 11 EP - 32 PB - Science Publishing Group SN - 2330-099X UR - https://doi.org/10.11648/j.sjc.20251301.12 AB - The first objective of the study is based on experimental characterization of the studied compound. The synthetization process of C11H8O4 (I) involved the O-acetylation of 6-hydroxycoumarin with acetic anhydride, utilizing diethyl ether as a solvent and pyridine as a base. The obtained structure was characterized by both spectroscopic analyses such as ESI-MS, FT-IR, 1H and 13C NMR analysis and by single-crystal X-ray diffraction studies. In the latter case, we employed direct methods to solve the structure of (I) and subsequently refined to a final R value of 0.054 for 1896 independent reflections. In the structure, C—H•••O hydrogen bonds connect the molecules into R22 (8) dimers, which are linked together by C—H•••O interactions, forming layers parallel to the bc crystallographic plane. Similarly, the crystal structure is sustained by π–π interactions between neighboring rings, with inter-centroid distances lower than 3.8 Å. The second objective of the study is to use theoretical calculation methods to analyze the effect of solvent polarity on the energy gap of the boundary molecular orbitals and the overall chemical reactivity of coumarin-6-yl acetate in order to provide a better understanding of stability and reactivity. A series of density functional theory computations were achieved with B3LYP/6-311++G(d,p) basis set in both gas and solvent phases. In addition to the dipole moment, the natural bond orbital charge distribution was estimated in toluene, tetrahydrofuran (THF) and benzene solvents. The calculations were conducted utilizing the Gaussian 09 software, and the outcomes exhibited that the solvents have an influence on the optimized parameters. Furthermore, dual and local reactivity indices as Fukui functions from the natural bond orbital (NBO) charges were estimated in order to have a better comprehension of the electrophilic and nucleophilic regions, as well as the chemical activity of (I). The obtained dipole moment in the gas phase is 6.03 Debye and those in the presence of the solvents are 7.89, 6.87, 7.51 and 6.83 Debye for water, toluene, THF and benzene, respectively. Additionally, the global chemical reactivity parameters exhibit variation contingent on the molecular compound and polarity of the solvents, making this an important consideration in the selection of appropriate solvents for a given chemical reaction. The studied compound shows higher stability in the benzene solvent evidenced by an EHOMO-ELUMO energy gap of 9.48 eV, while its low stability is observed in the gas phase with an EHOMO-ELUMO energy gap of 6.64 eV. VL - 13 IS - 1 ER -
Department of Training and Research in Electrical and Electronic Engineering, Research Team: Instrumentation, Image and Spectroscopy, Felix Houphouet-Boigny National Polytechnic Institute, Yamoussoukro, Côte d’Ivoire
Laboratory of Molecular Chemistry and Materials, Research Team: Organic Chemistry and Phytochemistry, University Joseph KI-ZERBO, Ouagadougou, Burkina Faso
Department of Training and Research in Electrical and Electronic Engineering, Research Team: Instrumentation, Image and Spectroscopy, Felix Houphouet-Boigny National Polytechnic Institute, Yamoussoukro, Côte d’Ivoire
Laboratory of Molecular Chemistry and Materials, Research Team: Organic Chemistry and Phytochemistry, University Joseph KI-ZERBO, Ouagadougou, Burkina Faso
Laboratory of Physics of Ionic and Molecular Interactions, National Center for Scientific Research, Aix-Marseille University, Marseille, France
Institute of Radical Chemistry, National Center for Scientific Research, Aix-Marseille University, Marseille, France
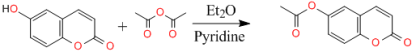
Figure 1. Synthesis of the title compound. Reagents and conditions: Pyridine, Diethyl ether, room temperature, 3 h.
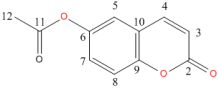
Figure 2. Numbering of carbon atoms used in spectra analysis.
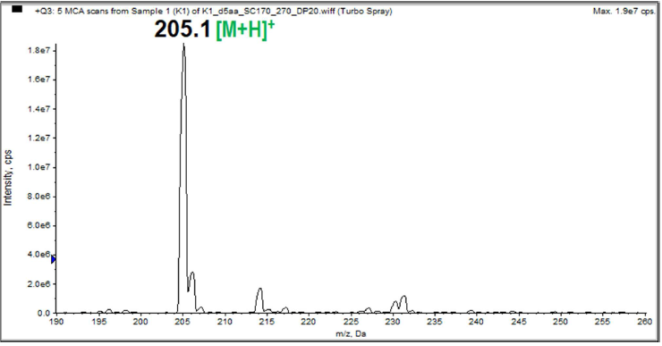
Figure 3. Electrospray ionization mass spectrum of the studied sample.
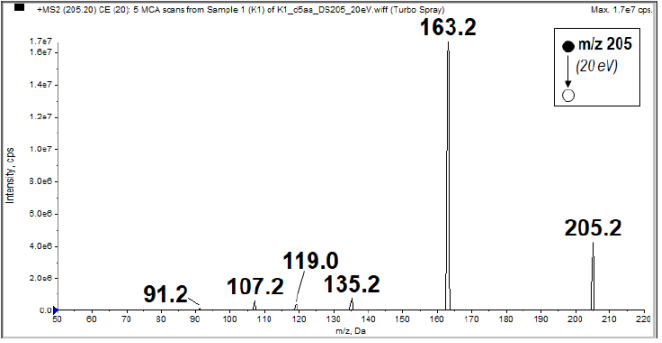
Figure 4. MS/MS spectrum of the protonated molecular ion peak (MH+) at m/z 205.
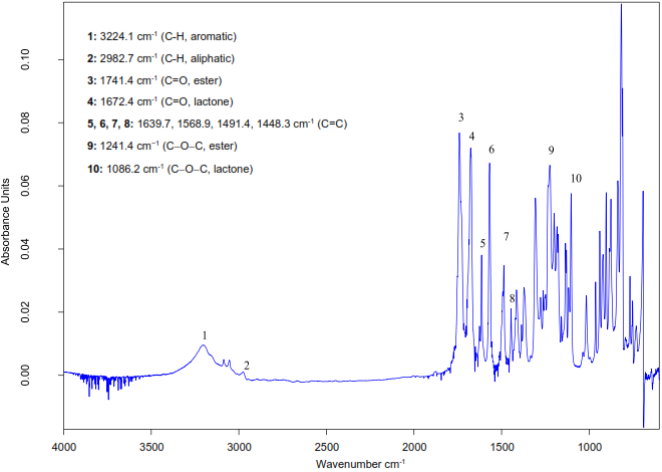
Figure 5. Experimental ATR-FTIR Spectrum.
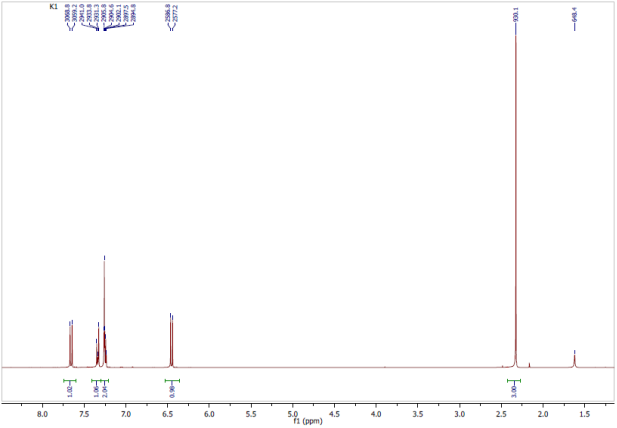
Figure 6. Experimental 1H-NMR Spectrum: CDCl3, 300 MHz.
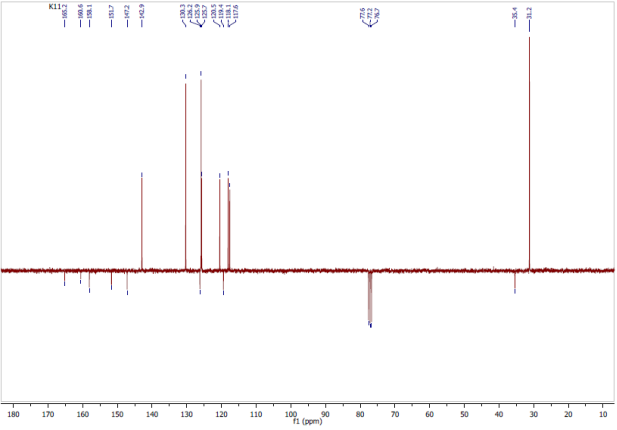
Figure 7. Experimental 13C (APT)-NMR Spectrum: CDCl3, 100 MHz.
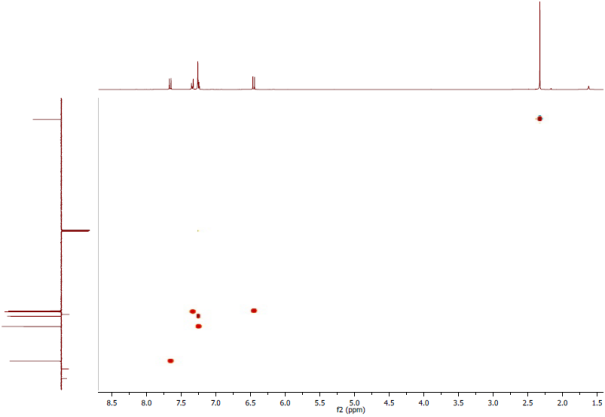
Figure 8. Experimental HSQC Spectrum: CDCl3, 1H-NMR 300 MHz; 3C (APT)-NMR 100 MHz.
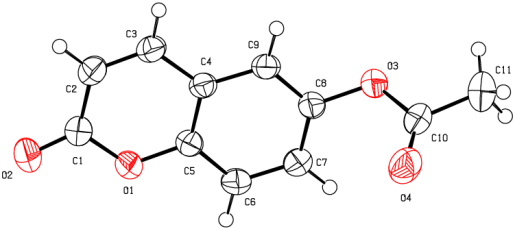
Figure 9. An ORTEP view of the title compound (I) with the atomic numbering scheme. Displacement ellipsoids are shown at the 50% probability level.
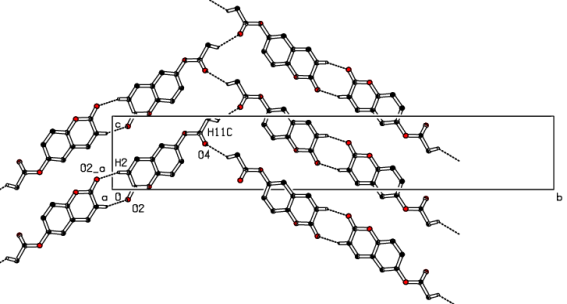
Figure 10. A view of the crystal packing, showing C—H···O hydrogen bonds linking molecules into dimeric units and their propagation into the bc plane.
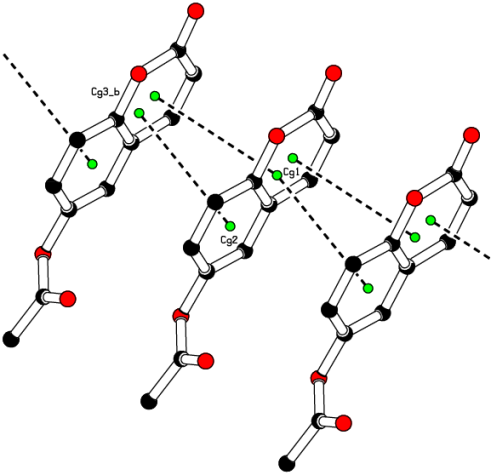
Figure 11. π···π stacking interactions in the crystal packing.
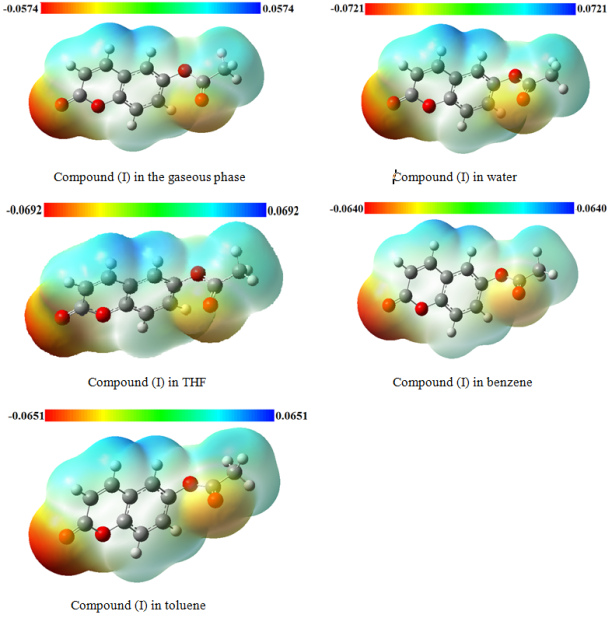
Figure 12. MEP map (in atomic units) calculated using DFT/ RM062X /6-311++G(d,p).
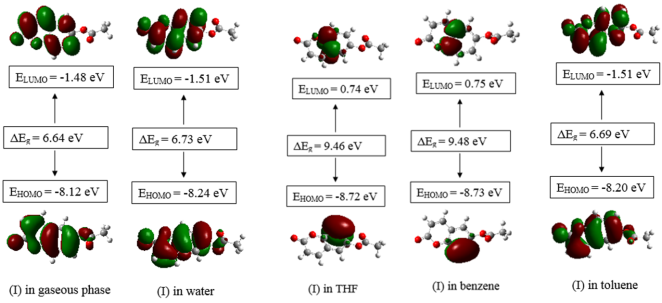
Figure 13. Calculated HOMO and LUMO orbital distributions and energy levels for the molecule (I) in solvents.
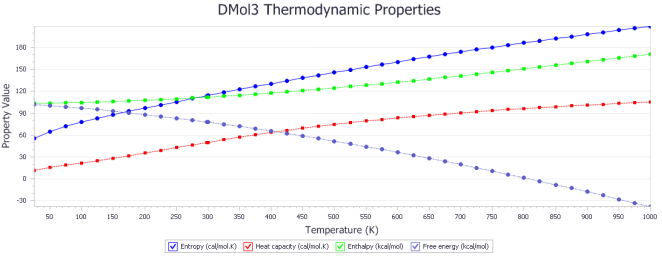
Figure 14. Thermodynamic property curves with entropy in cal/mol, heat capacity in cal/mol.K, enthalpy and free energy in kcal/mol.


 (8)
(8)